















.jpg)



Top Products
CPHI Pharmaceutical Products, Companies and News
.png)
News Drug Patent Expiries: a steep cliff or opportunity for innovation?
The pharmaceutical industry faces a patent cliff together in the years leading up to 2030. Learn what this means for drug pricing, their outsourcing partners, and drug innovation of the future.
.png)
News On Track at CPHI NA 2024: Supply Chain Economy with US Pharmacopeia
On the 7–9 May 2024 in Philadelphia, CPHI NA will be presenting an unmissable agenda, filled with content across 4 main tracks. In the following interview the Supply Chain Economy Track sponsor, the US Pharmcopeia introduces the track, and w...
.png)
News Pharma giants battle over COVID-19 patents in London courts
Two key players in the COVID-19 vaccine story – Pfizer/BioNTech and Moderna – have begun the latest chapter of their global legal battle over patents concerning technology for the development of mRNA therapeutics such as the COVID-19 vaccin...
Pharmaceutical Industry Content










Pharmaceutical Industry Webinars
-
.png)
Webinar Exploring Technological Trends in the Future of Pharmaceutical Manufacturing
-
23rd May 2024
-
4pm CET / 10am EST
-
-

Webinar Achieving Manufacturing Excellence Through Digital Transformation
-
16th April 2024
-
4pm CET / 10am EST
-
-
.png)
Webinar Made in Africa: What’s Driving Pharma Manufacturing
-
28th March 2024
-
4pm CET / 10am EST
-
-

Webinar Case Study: Risk Management for Annex 1 Sterile Production EMS
-
28th February 2024
-
4pm CET / 10am EST
-
-

Webinar Innovative Strategies for B2B Pharma Marketeers: Driving Value through Content
-
20th February 2024
-
4pm CET / 10am EST
-
-

Webinar Revolutionizing Pharma: Data and AI Unleashed
-
18th January 2024
-
4pm CET / 10am EST
-
-
.jpg)
Webinar Optimal Temperature: Elevating Biologics Cold Chain Excellence
-
16th January 2024
-
4pm CET / 10am EST
-
-

Webinar Market Outlook – The Biggest Pharma Trends of 2024
-
12th December 2023
-
4pm CET / 10am EST
-
-

Webinar The Next Frontier – Emerging Opportunities in the LATAM Pharma Market
-
21st November 2023
-
4pm CET / 10am EST
-
-

Webinar Vistamaxx™ MED - imagine the possibilities for healthcare product performance
-
10th October, 2023
-
4pm CET / 10am EST
-
-

Webinar Co-processing: A Multifaceted Approach for Enhancing Density & Powder Flow
-
19th September 2023
-
4pm CET / 10am EST
-
-
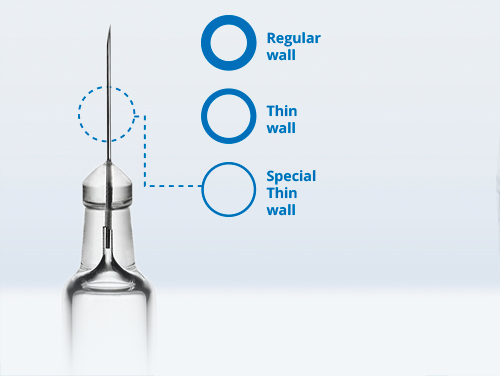
Webinar The Outlook for Cell & Gene Therapy Manufacturing
-
28th June 2023
-
4pm CET / 10am EST
-
-
Webinar Contract Packaging Outlook: Growth Trends within the Commercial Packaging Sector
-
25th May 2023
-
4pm CET / 10am EST
-
-

Webinar The HPAPI Boom: How Pharma is Rising to the Manufacturing Challenge
-
23rd February 2023
-
4pm CET / 10am EST
-




Sponsored Pharmaceutical Industry News
-
.png)
News The final six: meet the last Start-Ups exhibiting at CPHI NA 2024
The last six Start-Up companies that have made the list to exhibit in a dedicated area in Philadelphia at CPHI North America. Learn more about the companies and what makes them unique in the latest infographic. -

News The Patient-Centric Synergy of Pharmaceutical CDMO and CRO Collaborations
Pharmaceutical collaborations are nothing new to the industry. Increasingly complex drug development programs, calls for supply chain resiliency, and the involvement of all key stakeholders throughout a drug’s development lifecycle are pushing co... -

News Your Global Pharmaceutical Partner: Hangzhou Tomson Biopharma
Introducing TSN Biopharma -

News Asymchem is Positioned to be the CDMO of the Future
Why Asymchem is your most reliable CDMO partner -

News High demand: Vetter expands production capacities and services offered at its Austrian site
Pharma service provider further expands offerings for clinical development and manufacturing











-comp320105.png)


